PapersBiosecurityAI
Published
Author Stephen Turner
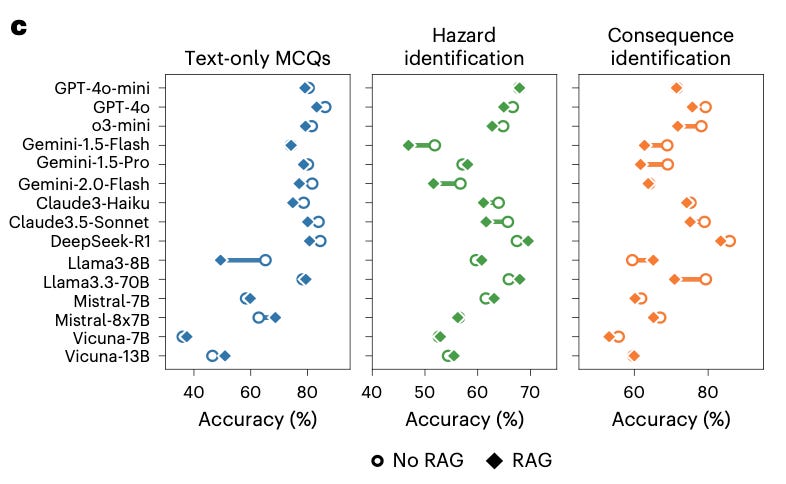
A laboratory safety benchmark finds retrieval augmented generation (RAG) can make strong models worse.
A laboratory safety benchmark finds retrieval augmented generation (RAG) can make strong models worse.
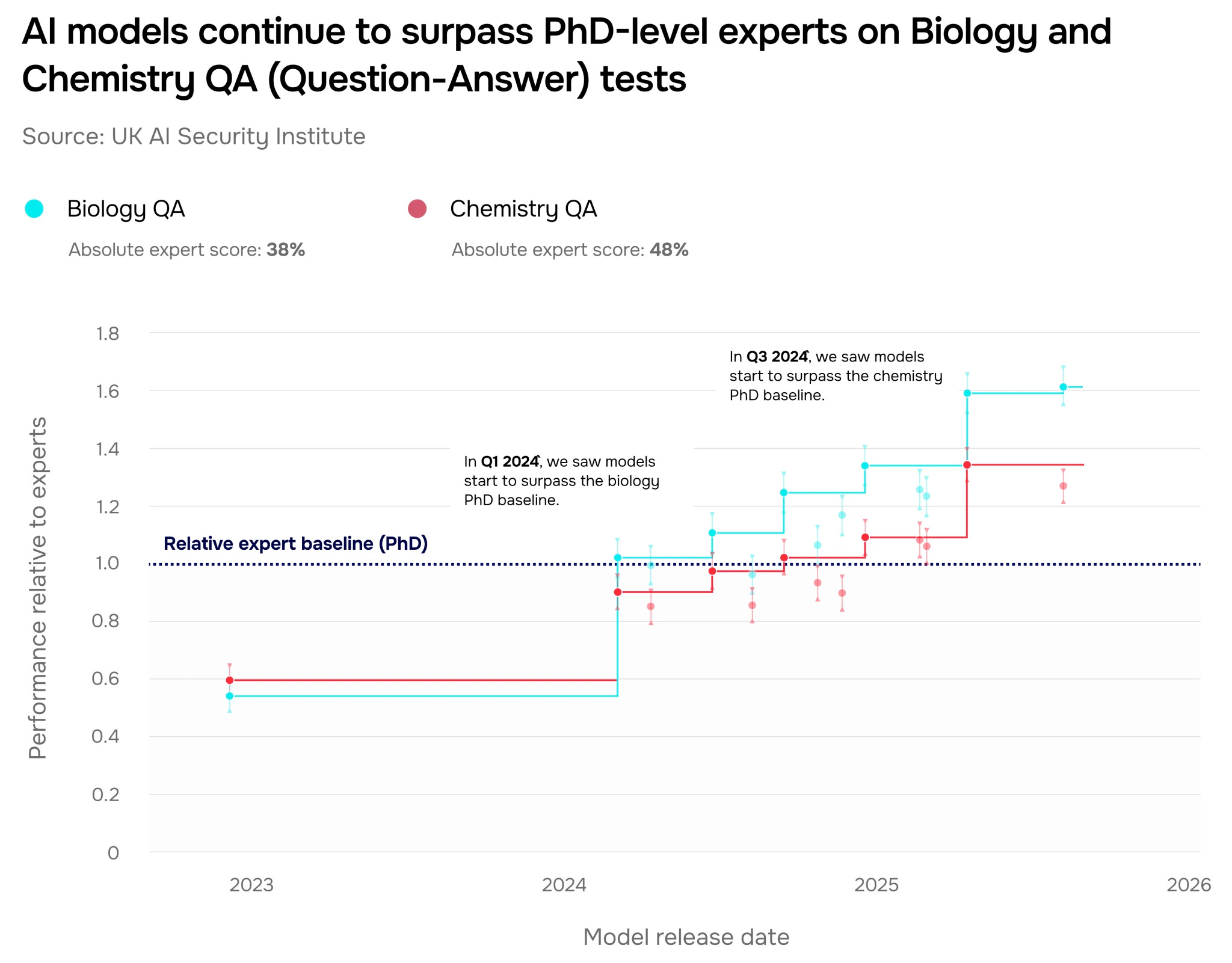
AIxBio, biotech 2025, Claude Code, Positron tips, cell fate engineering, academic slop, AI in health & life sciences research, R updates (R Data Scientist, R Weekly), RAG in R, papers &

AIxBio is here: Navigating the pacing problem and the future of global biosecurity
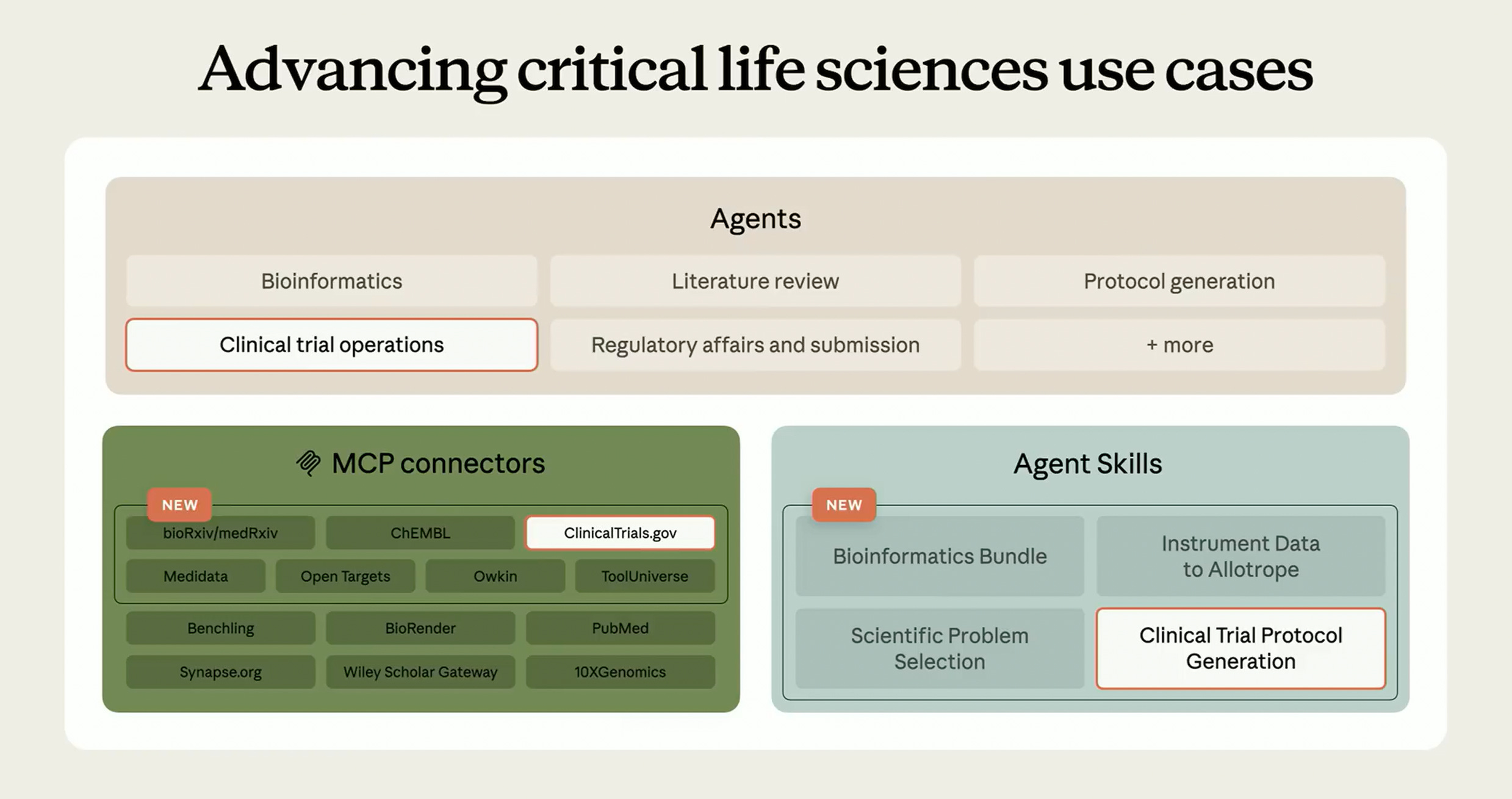
This week OpenAI and Anthropic launched their health/bio updates. While both companies are leaning into consumer-facing health concierges, Claude now has better tooling for life science researchers.

Reply to ‘Genome engineering for conservation might be a game changer but only with the incorporation of Indigenous voices’ in Nature Reviews Biodiversity

Not behind, but at the forefront: On feeling overwhelmed by AI progress, and why that means you're exactly where you need to be.
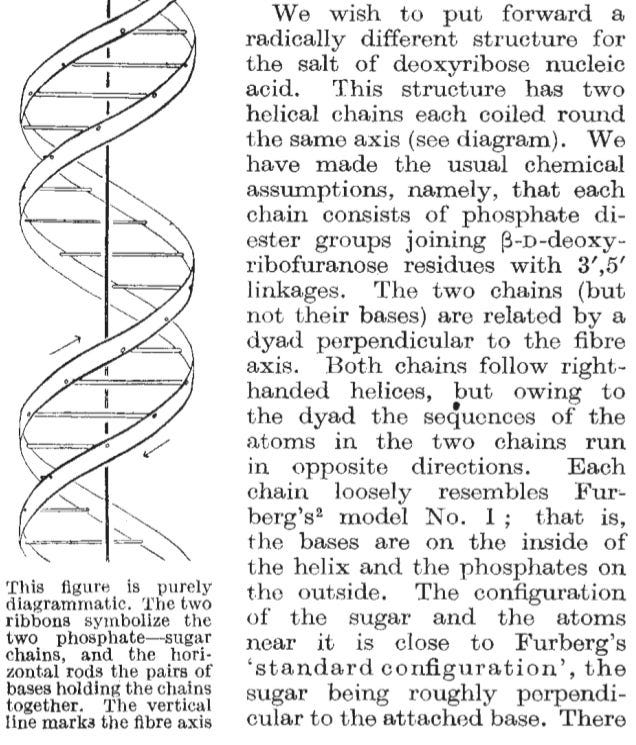
The science of responsible innovation, rv (uv for R), just quit, R updates (R Data Scientist, R Works), 5 things in biosecurity, why benchmarking is hard, open science (?), papers &
R + AI, uv, RAG+Zotero, Quarto books, Codex in Positron, Positron assistant &
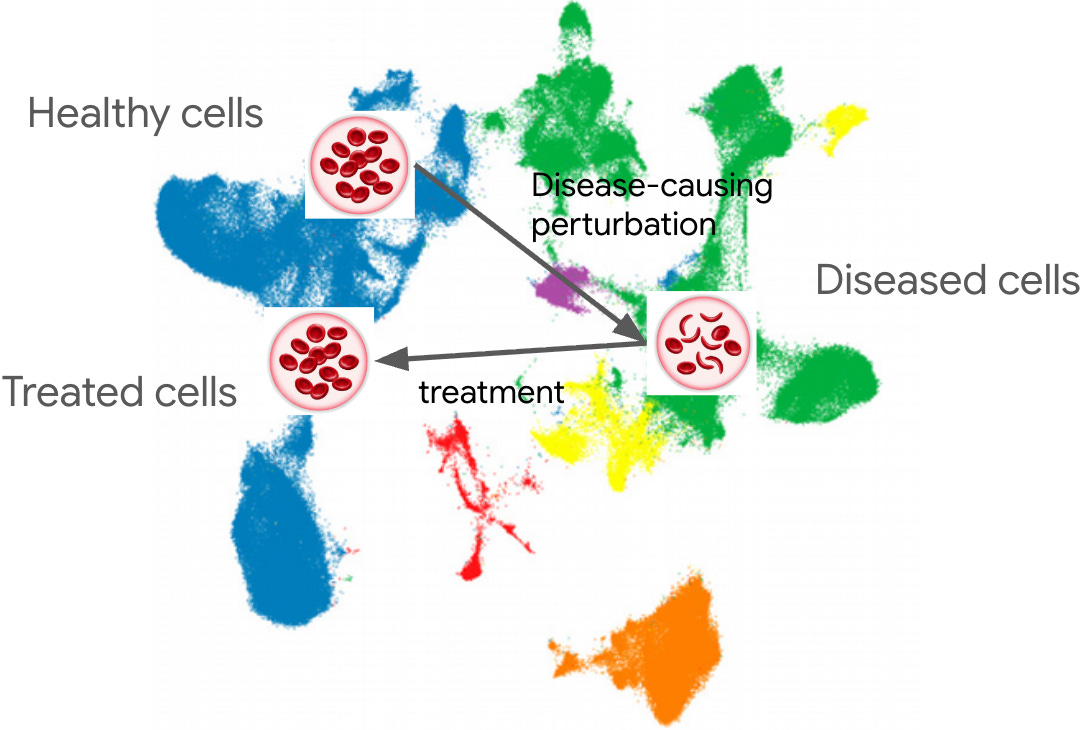
NSF reorg, Science in 2050, 2025 LLM recap, R+Python, R Data Scientist, virtual cells, genomics in 2026, Claude Code course, AI and labor, how uv got so fast, Anthropic/biotech, papers+preprints

Part 3 in a series of posts going back 14 years
On finding joy in slow, imperfect code in an age of copilots, agents, and chatbots. 1.2k words, 5 min reading time.