Interesting ChemistryCambridgeEnergyEnergy GapInteraction EnergyChemieEnglisch
Veröffentlicht
Autor Henry Rzepa

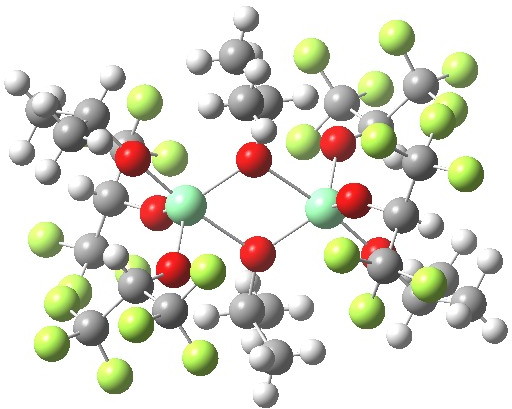
One frequently has to confront the question: will a hydrogen bond form between a suitable donor (lone pair or π) and an acceptor? One of the factors to be taken into consideration for hydrogen bonds which are part of a cycle is the ring size.